

时间:

作者:宋诗颖、崔潆心、凌霞、叶晓霞、乐健、许旭
他克莫司软膏浸没池体外释放评价方法的研究
摘 要
目的:建立他克莫司软膏体外释放评价方法,比较国内仿制制剂和参比制剂的体外释放行为一致性。
方法:采用A型浸没池建立他克莫司软膏的体外释放方法,选择无水乙醇-生理盐水(30∶70)为释放介质,孔径0.45μm的聚四氟乙烯膜为人工膜,转速200r·min-1。以液相色谱-串联质谱法测定释放量,采用Agilent Zorbax SB C18(150mm×2.1mm,3.5µm)色谱柱,以 0.1%甲酸溶液(含5mmol· L-1甲酸铵)(A)-甲醇溶液(B)为流动相,梯度洗脱,柱温55 ℃,进样体积10μL。测定他克莫司仿制制剂与参比制剂的释放量,并计算释放速率。采用基于Wilcoxon的秩检验和Mann-Whitney U检验的统计学方法计算仿制制剂与参比制剂的释放速率比值的90%标准置信区间,评价仿制与参比他克莫司软膏体外释放一致性。
结果:所建检测方法检测限达到0.026ng· mL-1,回收率在 92.6%~99.3%。2家国产他克莫司软膏仿制制剂中,仿制制剂1与参比制剂的释放速率比值的90%置信区间为97.92%~122.19%,在75.00%~133.33%范围内,显示体外释放速率与参比制剂一致,初、复试中仿制制剂2与参比制剂供试品释放速率比值的90% 置信区间均超出 75.00%~133.33%的范围,表明仿制制剂2的体外释放速率与参比制剂不一致。浸没池方法的研究结果与扩散池方法的研究结果一致。
结论:本文所建方法符合他克莫司软膏体外释放评价要求且结果可靠,浸没池体外释放法可用于他克莫司软膏的体外释放行为评价。
关 键 词

01 引 言
他克莫司是一种具有免疫作用的23元大环内酯类抗生素,能有效抑制T淋巴细胞的钙调神经磷酸酶激活。其相对分子质量较小,口服吸收能力差,可以制成半固体制剂局部用药治疗特异性皮炎、银屑病等皮肤病。他克莫司软膏是全世界首个获得批准上市的非皮质固醇激素类外用免疫抑制调节剂。目前,国内有十余家企业生产他克莫司软膏。
半固体制剂中活性成分体外释放的程度和速度受到原料药溶解度、粒径以及制剂的流变学性质等多个物理和化学参数的综合影响。外用半固体的体外释放试验(in vitro release test,IVRT)是表征和评价半固体制剂性能的有效手段,也用于药品开发过程中处方工艺的筛选研究。体外释放试验作为外用半固体制剂关键的体外质量研究项目,对于外用半固体仿制制剂质量研究与评价具有重要参考作用。《皮肤外用化学仿制药研究技术指导原则(试行)》 明确指出皮肤外用化学仿制药应开展体外释放研究。浸没池已用于环孢菌素眼用软膏、酮康唑乳膏的体外释放研究。褚信信等采用流通池开展复方克霉唑乳膏的体外释放研究。USP 2024推荐扩散池、浸没池用于半固体制剂体外释放研究,2020年版《中华人民共和国药典》暂无半固体制剂体外释放研究的相关要求和规定。扩散池是半固体制剂体外释放常用的方法。他克莫司软膏的体外释放研究多采用扩散池法进行,采用浸没池法开展他克莫司软膏的体外释放研究暂未见文献报道。
本文通过对释放条件的筛选建立他克莫司软膏浸没池体外释放测定方法,采用液相色谱- 串联质谱检测释放量,并完成相关方法学验证。以所建方法考察2家企业生产的他克莫司软膏仿制制剂与参比制剂的体外释放行为,并进行体外释放一致性评价,为他克莫司软膏质量的一致性评价提供新方法。本研究也将为浸没池法在其他外用半固体制剂体外释放评价中的应用提供经验。
02 仪器与试药
Agilent 1290-6490 超高效液相色谱三重四极杆串联质谱仪,Agilent 公司;DS-1206AT自动取样溶出系统及A 型浸没池,深圳市华溶分析仪器有限公司;0.45μm聚四氟乙烯亲水膜,龙津公司;Waters 2695高效液相色谱仪,Waters公司;透皮扩散仪(推荐使用华溶仪器TD-12AT Pro透皮扩散系统)。
他克莫司对照品(批号TAB0220803,纯度95.3%),海正药业;他克莫司软膏(参比制剂,批号C73736,规格10g∶1 0mg),安斯泰来制药有限公司;他克莫B司软膏(仿制制剂。T1∶A公司,规格10g∶10mg;T2∶公司,规格10g∶10mg);氯化钠(分析纯),国药集团化学试剂有限公司;乙醇(色谱纯)、甲酸(质谱级)、甲醇(色谱纯)、四氢呋喃(色谱纯),Merck 公司;甲酸铵(质谱级),CNW 公司。
03 方法与结果
3.1 浸没池体外释放
A型浸没池,详见图1。借助调整工具调节样品室体积以控制上样量。
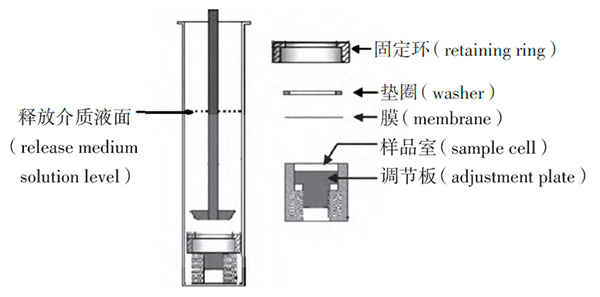
图1 浸没池结构(右)及其在溶出杯中(左)的示意图
采用A型浸没池与带有自动取样器的溶出仪,以预先超声脱气的无水乙醇-生理盐水(30∶70)100mL作为释放介质。取样品约0.50g,填充至浸没池样品室内,均匀涂布,精密称定,控制上样量偏差在±5%范围内。将人工膜覆于样品表面,用固定环固定后,将浸没池放入已装有释放介质的溶出杯中,转速 200r· min-1,水浴温度(32±0.5)℃,于0.5、1、2、3、4、5、6h时,分别取释放液3mL,并及时补充相同体积的释放介质。
3.2 色谱- 质谱条件
3.2.1 色谱条件
采用Agilent Zorbax SB C18(150 mm×2.1 mm,3.5 μm)色谱柱,以0.1%甲酸溶液(含5 mmol· L-1甲酸铵)(A)-甲醇溶液(B)为流动相,梯度洗脱(0~0.5 min,20%A;0.5~5.0min,20%A→5%A;5.0~7.0min,5%A→0%A;7.0~9.0min,0%A),流速0.3mL · min-1,柱温55 ℃,进样体积10μL,样品室温度6 ℃。
3.2.2 质谱条件
采用ESI源,正离子扫描模式,监测模式为多反应监测,以m/z 821.6 →768.5(碰撞电压为15V)为定量离子对,以m/z 821.6→786.6(碰撞电压为10V)为定性离子对。
3.3 溶液制备
3.3.1 对照品溶液
取他克莫司对照品约10mg,精密称定,置20mL量瓶中,加四氢呋喃适量使溶解并定量稀释至刻度,摇匀,制成每1mL中约含他克莫司0.5mg的溶液,作为储备液(4 ℃条件下,可放置1个月)。精密量取储备液适量,用无水乙醇-生理盐水(30∶70)逐步稀释制成每1mL中含5、10、50、100、150、200、250ng的溶液,作为对照品溶液。静置4h,待异构体转化平衡后进样。
3.3.2 供试品溶液
取他克莫司软膏体外释放试验中各时间点的释放液,即得。
3.4 方法学考察
3.4.1 线性与范围
取“3.3.1”项下系列浓度对照品溶液,按“3.2”项下条件进样测定,计算他克莫司峰面积(Y)与对应浓度(X)的线性方程Y=34 406X+182025 r=0.9996表明在5.12~255.80ng· mL-1浓度范围内,他克莫司的质谱响应与质量浓度之间线性关系良好。
3.4.2 进样精密度
取“3.3.1”项的5、50、100 ng· mL-1对照品溶液,分别连续进样6次,测定峰面积,他克莫司峰面积的RSD(n=6)分别为0.93%、1.1%和0.29%,均<2.0%,表明液质联用法进样精密度良好。
3.4.3 检测限和定量限
精密量取5ng·mL-1的对照品溶液,加入适量释放介质逐级稀释。按“3.2”项下条件测定,以信噪比为3时的浓度为检测限,以信噪比为10 时的浓度为定量限,他克莫司的检测限为 0.005 ng· mL-1(相当于最低释放浓度的0.03%),定量限为 0.026 ng· mL-1(相当于最低释放浓度的0.90%),灵敏度符合他克莫司软膏释放量测定要求。
3.4.4 回收率
取试验后所得释放液(质量浓度为121.14 ng· mL-1)1.0、2.0、5.0、5.0 mL,分别置于10mL量瓶中,再分别加入新配制的511.57 ng· mL-1他克莫司对照品储备液0.2、0.6、1.0、2.0 mL,用释放介质稀释至刻度,摇匀,作为20%、50%、100% 和150% 回收率溶液,一式3份。按“3.2”项下条件测定峰面积,计算回收率。回收率分别为96.7%(RSD=1.5%),86.2%(RSD=0.41%)、92.8%(RSD=1.1%)、96.2%(RSD=0.50%)。表明辅料/基质对样品的体外释放测定没有干扰。
3.4.5 溶液稳定性
取释放液3份于(34±0.5) ℃的水浴条件下放置16、24h,按“3.2”项下条件测定释放液放置前后他克莫司的浓度。放置16、24h后他克莫司浓度与初始浓度比值分别为101.3%、100.4%,表明供试品溶液在34 ℃的温度条件下,24h内稳定。取对照品溶液于6 ℃下放置168h,按“3.2”项下条件测定,168h后他克莫司浓度与初始浓度比值为101.52%,表明对照品溶液在6 ℃条件下168h内稳定。
3.4.6 释放精密度
分别取参比制剂样品约0.50g,一式6份,精密称定,按“3.1”项下方法及“3.2”项下条件测定累积释放速率。连续2d重复测定,得到12组累计释放量结果。12条拟合直线相关系数r均>0.94,释放速率为10.3%,日内精密度为8.4%,日间精密度为9.0%,RSD 均<10%。因此,建立的测定方法符合他克莫司软膏的体外释放测定的精密度要求。
3.4.7 物料平衡
取试验结束后的剩余膏体与人工膜(共6组),用加热至60 ℃的四氢呋喃20mL分次将残留软膏溶解并转移至50 mL量瓶中,用加热至60 ℃的四氢呋喃10mL分次淋洗垫片、浸没池样品室部分、人工膜,合并洗液至同一50 mL量瓶中,加水10mL,小心振摇成混悬液,用无水乙醇稀释至刻度,剧烈振摇,冷藏30min,4000r· min-1离心10min,精密量取上清液1mL,置50mL量瓶中,用释放介质稀释至刻度,摇匀。精密量取1mL置10mL量瓶中,用释放介质稀释至刻度,摇匀,作为残留膏体供试液。将残留膏体供试液按“3.2”项下条件测定峰面积,计算膏体中他克莫司残留量。膏体中残留量及累积释放量二者之和与初始上样量相比较,计算物料平衡。6组结果分别为96.16%、95.12%、92.56%、93.45%、99.27%和96.17%,均在90%~110%的范围内,表明实验过程中,他克莫司未出现物料流失,结果可靠。
3.5 样品测定
取仿制制剂与参比制剂,按“3.1”项下方法制备释放液,按“3.2”项下条件测定释放曲线(仿制制剂与参比制剂交替上样),仿制制剂与参比制剂的单位面积累积释放曲线见图2、3。
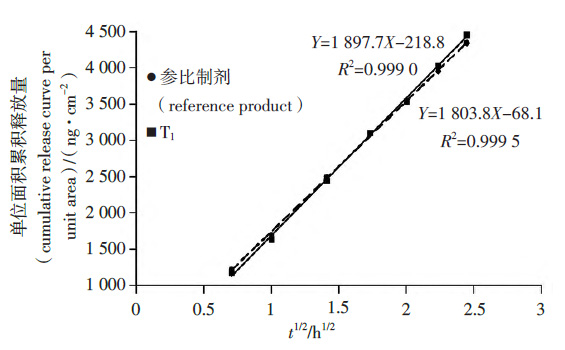
图2 参比制剂与T1 的单位面积累积释放曲线
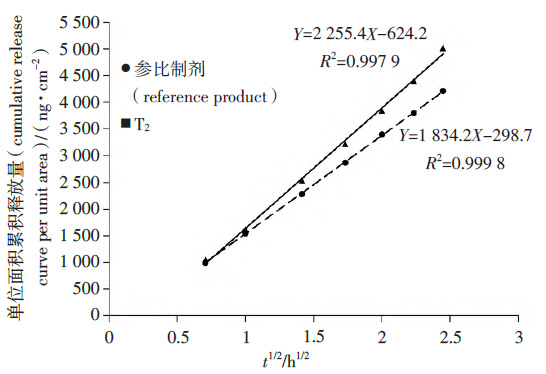
图3 参比制剂与T2 的单位面积累积释放曲线
仿制制剂与参比制剂的一致性采用基于Wilcoxon的秩检验和Mann-Whitney U检验统计学方法进行。求出仿制制剂与参比制剂释放速率中位数差异90%置信区间,然后进行一致性判断。
分别计算T1与参比制剂的释放速率,并计算二者的比值,结果见表1。
表1 T1 与参比制剂的体外释放速率比值
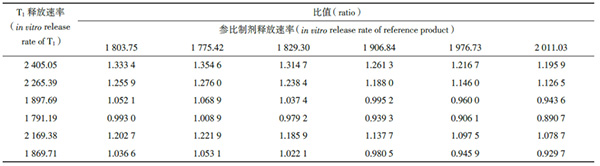
将表1中36个比值按升序排列,第8个和第29个比值分别是T1的中位体外释放率(斜率)与参比制剂的中位体外释放率比值的90% 置信区间的下限和上限,本实验中90%置信区间为97.92%~122.19%,在75%~133.33% 范围内,表明T1与参比制剂的释放速率等同。
在T2与参比制剂的对比实验中,初试的90% 置信区间为115.10%~152.89%,不在75%~133.33%范围内。重复进行2次试验,增加T2与参比制剂各12个斜率,2种样品各18个斜率进行释放速率比值计算。
324个数据进行升序排列,位于第110位和第215位的比值分别是T2的中位体外释放率与参比制剂的中位体外释放比率比值的90%置信区间的下限和上限。复试试验中90%置信区间为115.87%~135.29%,仍然不在75%~133.33%,表明T2 与参比制剂的释放速率不等同。
04 分析与讨论
4.1 检测方法选择
他克莫司软膏的规格小,含量仅为0.1%,且释放量低,他克莫司HPLC紫外检测的检测限约为0.1μg,难以满足释放量测定要求。因此采用灵敏度更高的三重四极杆质谱检测器进行他克莫司释放量的测定。
4.2 人工膜的选择
从对药物无阻滞作用与无吸附作用2个方面进行考虑。选用0.45μm尼龙膜、0.45μm聚四氟乙烯亲水膜(PTFE 亲水膜,光滑面接触膏体)和0.45μm聚四氟乙烯疏水膜(PTFE疏水膜,光滑面接触膏体),分别取参比制剂样品0.50g,按“3.1”项下方法及“3.2”项下条件测试。PTFE亲水膜、PTFE疏水膜、尼龙膜6h释放液浓度分别为1.0121、0.3502、0.8752μg·mL-1,表明PTFE亲水膜对药物的阻滞性最小。
取“3.3.1”项下配制的100ng· mL-1对照品溶液于试管中,分别加入0.45μm尼龙膜和0.45μmPTFE亲水膜,放置24h后,轻取上层清液,按“3.2”项下方法测定峰面积,计算24h后溶液中他克莫司含量的变化。结果PTFE亲水膜和尼龙膜释放液与对照原液浓度比值分别为97.8%、92.3%,表明PTFE亲水膜对他克莫司的吸附性最弱。
对膜正反面的阻滞性差异进行研究。以PTFE亲水膜的不同面接触样品表面,使用垫片与固定环固定。按“3.1”项下方法及“3.2”项下条件测定累积释放量并绘制释放曲线,见图4。显示光滑面接触软膏时,PTFE 亲水膜对释放的阻滞效用最弱。
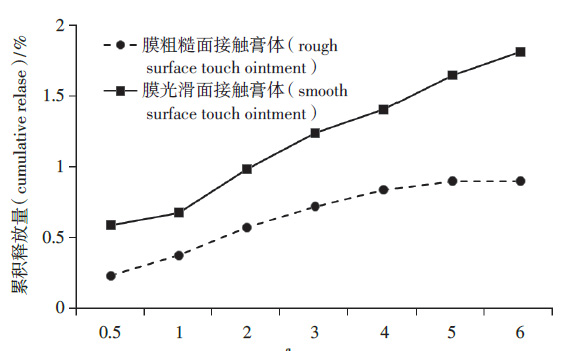
图4 PTFE 亲水膜正反面比较
综合以上结果,选择0.45μm PTFE 亲水膜为人工膜,且使用时光滑面朝下接触膏体。
人工膜的选择是体外释放方法建立的重要步骤,理想状态下人工膜对软膏应仅具支撑作用,对药物释放没有阻滞影响。常见的PTFE膜因氟原子极化率低和结构特性,持久亲水性不强。本研究所选用的PTFE亲水膜采用亲水改性方法使得PTFE膜正反两面表观不一致,正面有涂层,表面平整光滑,反面没有涂层,表面粗糙,肉眼可见纤维纵横交错。实验中发现反面(粗糙面)接触软膏时,膜对他克莫司的释放有明显的阻滞作用。可能是由于粗糙面接触软膏时交错的疏水纤维容易形成微小空隙,释放过程中空隙内残留的空气影响介质和膏体的充分接触,从而表现出单向阻滞性。而亲水改性的正面(光滑面)接触膏体时,介质充分接触膏体,保证了药物的释放。
4.3 释放介质的选择
释放试验中的释放介质应模拟实际生理条件,如生理盐水、磷酸盐缓冲液(pH 7.4)等。由于他克莫司在磷酸盐缓冲液中不稳定,因此选用生理盐水。他克莫司难溶于水,在水溶液中溶解度低,因此在释放介质中加入一定量无水乙醇提高他克莫司软膏在释放介质中的释放量。试验发现,当释放介质中有机相比例提升,他克莫司软膏的释放量增加,但当无水乙醇- 生理盐水的比例达到40∶60时,他克莫司降解产生杂质,影响释放量检测。
漏槽条件是体外释放试验必须满足的条件之一,药物在释放液中的溶解度应为释放液中药物最高浓度的3~5倍。经检测他克莫司在无水乙醇- 生理盐水(30∶70)中的溶解度为1.44 mg· mL-1,实验中释放液的最大质量浓度为115 ng· mL-1,满足漏槽条件。
综上结果,选择无水乙醇-生理盐水(30 ∶ 70)100 mL作为释放介质。
4.4 转速的选择
按“3.1”项下方法,比较150和200r· min-1 2种转速的差异。2种转速下,6h测得释放液质量浓度分别为0.0849和0.1037μg· mL-1,表明当转速较高时,他克莫司软膏的释放速率更快。
4.5 取样时间的选择
按“3.1”项下方法,于0.5、1、2、3、4、5、6、9、12、15与18h取样,按“3.2”项下条件测定峰面积,计算累积释放量并绘制释放曲线,结果显示他克莫司软膏释放缓慢,18h累计释放量仅有2.8%。经计算,释放过程符合Higuchi方程。释放曲线线性部分应至少取5~6个时间点,因此确定取样时间点分别为0.5、1、2、3、4、5 和6h。
4.6 与扩散池法的对比
与实验室前期建立的他克莫司软膏Franz扩散池体外释放测定法进行对比验证。
采用改良的Franz立式扩散池进行测定,以无水乙醇- 生理盐水(30∶70)12 mL 作为释放介质,搅拌速度 400 r· min-1,介质温度(32±0.5)℃,经0.5、1、2、3、4、5和6h时取释放液4mL,取样后补充相同体积的释放介质。
精密称取他克莫司软膏0.3g,放置于垫有PTFE亲水膜的2mm厚定量环中,均匀涂布并保证表面平整。将完成称量的定量环放置并固定在扩散池上,依照上述条件试验(仿制制剂与参比制剂交替进样),各时间点释放液即为供试品溶液。
精密量取“3.3.1”项下他克莫司储备液适量,用释放介质定量稀释为每1mL中含他克莫司0.5μg的对照品溶液。静置4h,待异构体转化平衡后进样。
采用十八烷基硅烷键合硅胶为填充剂(Agilent Zorbax SB色谱柱,150mm×4.6mm,5μm),以水-异丙醇- 四氢呋喃(5∶2∶2)为流动相,检测波长为220nm,流速1.0mL · min-1,进样体积100μL,样品室温度5 ℃,柱温60 ℃。理论板数按他克莫司峰计算应不低于2 000。精密量取对照品溶液与供试品溶液各100μL进样测定。
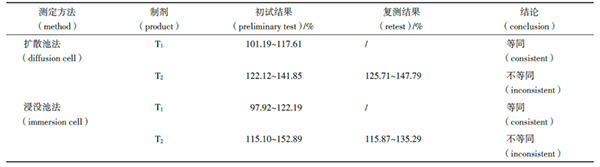
按上述试验条件进行参比制剂与T1、T2的测定。2种方法测定结果汇总如表2,2种释放测定方法的等效性评价结果一致。
表2 不同体外释放方法结果对比
05 结 论
体外释放试验可以反映半固体制剂中药物的微观结构与流变学性质等理化参数的综合作用,适用于多种场景。对于仿制制剂而言,可用于评估与参比制剂间的一致性,作为仿制制剂的生物等效性研究豁免依据之一。本研究针对他克莫司软膏体外释放评价方法的建立与应用展开研究,建立了他克莫司软膏的体外释放浸没池评价方法。方法学验证结果表明所建方法具有良好的区分力,结果准确,可用于他克莫司软膏的体外释放一致性评价。将所建方法应用于不同企业生产的仿制制剂与参比制剂的一致性评价。经试验研究与统计学分析,T1与参比制剂的释放速率等同;而T2与参比制剂的释放速率不等同。
扩散池法是目前应用较多的半固体制剂体外释放评价方法,浸没池的相关文献则较少。本研究将浸没池法与扩散池法评价结果进行对比验证,结果表明,2种释放方法的一致性评价结果相同。本研究所建立的浸没池法评价方法可用于他克莫司软膏的体外释放评价。
实验中发现浸没池的方法精密度略低于扩散池法的精密度,可能与浸没池装置本身结构较为复杂以及影响因素较多有关,如不同浸没池之间的系统误差、浸没池的密封性等,有待于在实验中不断认识和改进。
本研究为他克莫司软膏的质量一致性评价提供了新的体外释放评价方法,并为半固体仿制制剂的体外释放试验的装置选择积累了数据与经验。
06 参考文献
略
07 溶出度与释放度测定法




华溶五大溶出系统产品











